
의료시장정보
미국"보건의료개혁이 한국 제약,바이오업계에 미치는 영향"
| 작성자 | 관리자 | ||
|---|---|---|---|
| 등록일 | 2012-07-13 | 조회수 | 8,531 |
| 출처 | KHIDI | ||
| 원문링크 | - | ||
| 첨부파일 | |||
1. 보건의료개혁이 한국 제약, 바이오 업계에 미치는 영향
□ 의약품 수요증가에 따른 글로벌 제휴협력 가능성 증대
○ 미국의 건보개혁으로 전 국민의 95%이상이 의료혜택을 받게 되는 등 의약품의 대규모 수요 증가 예상
- 1,600만 명 규모의 실질 신규 민영보험 가입자 발생, 의약품 가격의 합리화와 비용에 따른 의약품 사용주안 요인 제거 등 의약품 사용촉진 환경 형성
- 세계 제약시장의 40% 이상을 차지하고 있는 미국 의약품 시장의 이 같은 지각변동은 산업흐름 네트워크를 따라 글로벌 제약시장으로 파급 예상
(1) 기회 1 : 글로벌 라이센싱
○ 반면, 개혁 초기 제약업계에 부과된 비용부담, 파이프라인의 고갈과 연구개발의 생산성 위기(R&D Productivity Crisis) 등 제약 산업이 갖는 고비용·고위험 구조로 인해 글로벌 수준의 제휴협력 가능성 증대 예상
- 현재 제약기업 간 및 제약-바이오기업 간 인수합병, 기업 간 라이센싱을 통한 파이프라인 확보가 활발히 이루어지고 있고,
- 신약후보물질에 대한 수요가 꾸준히 증가하고 있는 등 다양한 내외적 환경의 영향으로 기업 간 제휴협력의 필요성 최고조
○ 세계 제약 산업의 선도 55개 기업을 중심으로 한 라이센싱인 제품의 비중 분석에서 선도 55대 제약사의 기술구매 의존도가 지속도로 상승
<그림> 연도별 55대 제약사의 처방약 판매액 중 라이센싱 인(Licensing In) 제품 비중
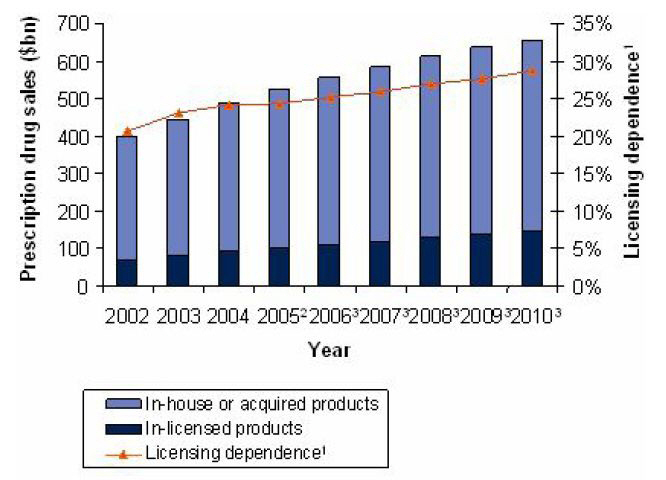
Source : Data Monitor, 2007
· 다국적 제약사 브리스톨마이어스(BM)의 경유, 바이오의약품 파이프라인 확보 및 바이오 기업인수를 통해 스스로 바이오제약기업으로의 변환을 모색한다고 밝힘(2010년 1/4분기 경영실적 보고에 따르면 현재 약 100억 달러 정도의 유동성을 보유하고 유망한 제품군 확보 추진 중)
· Eisai, Daichii Sankyo 등 일본 제약업계는 미국 거대제약사에 라이센싱하는 형식으로 11개 회사가 50대 제약사로 성장하고 매출액의 40~50%가 미국 등 세계시장에서 발생
· 미국 뉴저지에 위치한 Forest Laboratories는 Biovail사의 Tiazac, Lundbeck사의 Celexa를 Licensing-in하여 브랜드 업체로 성장. 본 기업은 임상 2상 이전의 초기단계 개발은 없이 라이센싱에만 의존하는 모델로 비약적 성장
○ 한편, 국내 제약사 및 바이오기업들 역시 글로벌 라이센싱을 위한 다양한 기술개발이 가시적인 단계까지 성장
- 반명 국내 제약사들은 글로벌 임상 및 마케팅 능력을 보유하고 있지 못하고 임상 2상 이후의 개발비용을 부담할 재정적 능력이 없어 라이센싱 아웃을 통한 기술이전이 매우 유력한 모델로 자리매김할 전망
○ 여기에 정부주도의 의약품 기술 수출입지원이 시너지 요인으로 작용할 전망
- 우리나라 정부는 ‘제약산업 경쟁력 강화방안’ (9개 부처 합동, 2010.2)을 통한 체질개선 등 글로벌 경쟁력 강화를 목표로 기술이전 활동 등에 대한 장려 정책 추진하고 있음
- 국내 우수 제약기술의 연구현황, 기술내용, 연구자 정보 등에 대한 DB 구축 및 기술평가를 통해 기술 수출 활성화를 촉진하고 있으며,
- 해외마케팅용 영문기술 작성 지원 및 국내외 기술수출입 상담회 (licensing partnership) 개최를 통해 기술 수출입 촉진
- 오프라인 기술마케팅을 위해 한국보건산업진흥원 등 보건의료 전문기관을 통해 BIO KOREA를 연례 개최하고 있음
· BIO KOREA 2009(‘09.9.16~18)의 경우 해외기업 38개사 참여 파트너링 실시(257건 상담)
○ 그러나, 지난 20년간 우리 제약산업의 글로벌 라이센싱 아웃 실적은 현재까지 (89~07년 현재) 총 40여건 수준으로 조사 (보건산업진흥원, 2007)
- 미국시장 진출은 총 10여 건으로 집계되어 성과를 제고하는 것이 과제
○ 따라서, 신약개발 라이센싱 관련 수요 정보의 신규성 확보와 개발기술의 품질제고에 주력하고, 미국 시장의 기술이전 트렌드에 대한 업데이트가 주요한 과제임
- 글로벌 기업의 제휴 수요 부분 및 제휴모델 분석, 제휴사의 경쟁력 평가항목 등에 대한 분석이 필요
- 또한 질환군에 따라서 기술이전(licensing-out)이 가능한 개발단계(stage)가 다를 수 있어 Indication별 전략수립이 필요
· 일부는 phase 26 이상 요구, 일부는 임상 1상 단계에서도 deal 가능
○ 지금까지 다국적기업과 대형 벤처캐피탈사가 집중적인 투자를 해오지 않은 초기개발단계(early stage)에 상대적으로 저평가된 좋은 기술들이 많이 있어, 이런 기술들의 효율적 개발시도가 글로벌 트렌드화 하고 있음
- 초기단계(early stage)기술 보유가 많은 국내 바이오기업들에게 이러한 트렌드 변화는 새로운 기회로 기능할 수 있음
- 잘하는 기술에 집중하지 말고 필요한 기술에 집중하여, 연구개발 단계부터 상용화 (commercialization)를 고려하는 관점 수립 필요
○ 현재 미국에서 진행 중인 기술이전의 동향과 수요는 메드트랙(Medtrack)등 유료 데이터베이스(www.medtrack.com/research/default.asp)를 통해 실시간으로 확인할 수 있음
○ 미국 주요 제약사의 기술이전 트렌드는 대량생산의 블록버스터 의약품이 아닌 높은 바이오 기술력에 대한 니즈로 상대적으로 경쟁이 적은 희귀질환(orphan disease)분야로 빠르게 전환되고 있음
<그림> 상위 10대 기업 기술이전 트렌드가 니치마켓으로 변화 (2009년 vs 2008년)
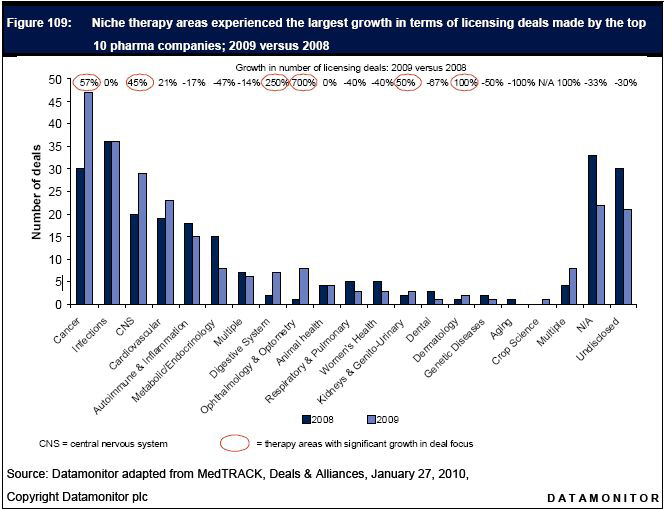
- GSK의 경우 2010년 2월 경영공시를 통해 전통적인 항우울제 및 진통제에 대한 연구를 중단하고 알츠하이머와 파킨스씨 병에 대한 연구에 집중한다고 밝힌 바 있음
- 이 같은 변화가 촉발되는 요인은 대규모 임상이 불필요함에 따른 비용과 기간의 감소, 대규모 판매부서 및 유통채널의 불필요 등 비용을 절약할 수 있는 요인이 많은데다 정부의 신속한 승인과 각종 혜택 부과 등에 의해 성공률이 높기 때문임
· 실제로 지난 2000년부터 2002년까지 희귀질환 의약품(orphan drug)으로 인정된 사례는 207건이었으나, 2008년부터 2010년까지는 444건으로(FDA 2010) 114% 성장했으며, FDA에 의한 희귀질환 의약품 승인도 같은 기간 32개에서 47개로 무려 46% 성장
- 이 같은 기술이전 트렌드에 대한 신규성 유지가 필수적임
(2) 기회 2 : 글로벌 아웃소싱
○ 라이센싱이 신약개발의 필수 요소로 자리매김함과 동시에 신약탐색 및 발굴과정과 임상시험 관리과정 등 신약개발의 일정단계를 아웃소싱 하는 글로벌 아웃소싱 역시 중요도가 점차 확대되고 있음
○ 또한 글로벌 기업에 대한 아웃소싱 부문에서는 임상연구에 활용되는 임상용 의약품의 제조, 분석, 원료, 완제 품질관리자료(CMC) 등 용역분야도 기회 영역으로 포함 가능
- 다국적 임상시험의 국내 유치를 통한 공급 계약 확보 역시 가능
· 임상시험의 경우 우리나라는 지난 2000년 ICH-GCP를 도입하고, 2007년 국가임상시험사업단(KONECT)을 설치한데 힘입어, 2000년 3건에 불과하던 다국적 임상(multinational clinical trials)이 2008년에는 216건으로 72배 성장 (Source : Journal For Clinical Studies, Dohyun Cho, November, 2009)
· 국내 기업 중 미국 내에서 FDA 승인을 받고 주요 미국 제약사에 원료합성, 판매 등을 규모 있게 하고 있는 기업으로는 유한화학이 있으며, cGMP 수준의 바이오의약품 생산시설을 가진 셀트리오, 지도부딘, Antiviral 등 중간체로 특화된 삼천리제약(동아제약에 인수) 및 SK 생명과학 등도 중간체 매출 비중이 높음
· 지방에 위치한 소규모 기업인 한국화인케미컬(Korea Fine Chemical) 역시 cGMP 기준으로 제품을 생산하고, 중소규모의 동국제약 역시 고난도 기술과 자본집약이 필요한 프로포폴을 유럽의약청(EMEA) 승인을 받아 유럽의 유수 기업에 납품. SK 역시 오페프라졸을 독일에 안정적으로 납품
· 아웃소싱에 성공한 기업들은 일반적으로 내수시장을 타깃으로 한 바가 없고 품질관리에 많은 노력을 기울여 왔음
○ 완제 및 원료의약품(API) 생상의 글로벌 아웃소싱 역시 확대될 전망
- 특히 우리나가 강점을 가지고 있는 중간체의 생산과 원료의약품 생산, 승인된 의약품(Authorized Drug)의 위탁생산 등에 관심 있는 관찰이 필요 됨
- 특히 메이저 제약사들의 제품군을 위탁생산하기 위해서는 신뢰관계 구축이 선행되어야 함
○ 제네릭의약품의 경우는 현재 미국 FDA에서 승인되는 완제 및 원료의약품의 81%가 중국과 인도 2개 국가에서 생산
- 미국 식품의약품국(FDA)의 제네릭의약품 승인신청 생산시설 소재지 현황에 따르면, 2007년 기준 총 신청건수 1,154개 중 미국 소재는 13%에 불과 (중국 소재 43%, 인도 소재 39%)
- 인도는 저렴한 가격경쟁력과 더불어 공장시설을 미FDA가 인증하는 cGMP 기준1)으로 밸리데이션(Validation)2)까지 의무화하는 등 품질제고 노력을 통하여 복제약 수출 강국으로 부상
○ 그러나, 가격경쟁력으로 시장을 점유해 오던 상기 2개국의 생산시설 및 원료의약품(API) 등에 대한 품질문제가 주요한 이슈로 부각
- 특히, 미국 내 대형 제약기업들이 중국과 인도에서 공급받는 원료의약품(API) 생산시설의 제조품질관리기준에 대한 우려가 확산되고 있음
· 미국 FDA는 2008년 수술에 반드시 필요한 혈액응고제 헤파린(heparin)의 중국 원료생산 공장을 품질을 이유로 전면 중단시킨 사례가 있으며 유사한 우려가 지속적으로 제기
- 품질문제의 제기는 수요확대와 더불어 전면적으로 부상될 여지가 높으며, 현재 투명성이 보장되지 않고 있는 원료의약품 공급처 공개 등도 제도화압력이 있음
· 제약기업들은 보통의 약품의 공급체인을 영업비밀로 분류하고 있기 때문에 실제 의약품 원료의 공급처 파악은 매우 어려움. FDA가 의약품 원료 승인규정(DMF, Drug Master Files)을 통해 의약품 공급처를 공개적으로 리스팅하고 있으나, 제약기업이 원료공급사의 정보를 게재하는 것이 의무사항이 아니기 때문에 공급처와 관련된 내용의 신뢰도는 매우 낮음.
<표> 중국과 인도 생산공장의 최신 미국 FDA 적발 사례
|
국가 |
날짜 |
적발기업 |
조치내용 |
적발사유 |
|
중국 |
2010.1.28 |
Xian Libang제약 |
Warning Letter |
cGMP 위반 등 |
|
2009.4.14 |
Shanghai No.1 제약 |
Warning Letter |
허위서류 등 | |
|
2009.4.14 |
Qingdao Jiulong 제약 |
Warning Letter |
contamination 등 | |
|
인도 |
2008.9.16 |
Ranbaxy 제약 |
Warning Letter, Import Alert |
cGMP시설 미증축 |
Source : 보건산업진흥원 뉴욕지소 요약, 2010
○ 따라서, 국내 생산 완제의약품 및 원료의약품(API) 품질에 대한 우수성이 확보되면 미국시장 진입 가능성이 높아질 수 있음
- 그러나 이를 위해서는 반드시 엄격한 품질 확보가 진행되어야 함
- 의약품 품질관리기준 선진화 및 원활한 도입을 위해 우리나라 식품의약품안전청에서도 2010년 1월 Validation 등을 전면 실시
· 품목별 사전 GMP평가제 및 Vaildation 의무화 : 신약(‘08.1월) → 전문의약품(’08.7월) → 일반의약품(‘09.7월) →기타(’10.1월)
○ 글로벌 라이센싱에 대한 상시적인 수요모색 및 파트너 확보 활동 필요
□ 영제네릭의약품 진출 가능성 확대
○ 현재 전세계 제네릭시장의 규모는 연간 800억 불에 달하고 있으며, 미국은 이중 과반수에 달하는 420억 불 시장규모임
· 제네릭 시장규모의 연간 성장률은 8.2%로 시장은 더욱 커질 전망(IMS Health, 2009)
○ 이 같은 제네릭의약품의 증가세는 블록버스터 의약품들의 특허권 만료(2011~2013)가 집중되는 시기적 환경에 의해 더욱 탄력을 받을 것으로 예상
· Prevacid(lansoprazloe), Topamax(topiramate), Valtrex(valacyclovier) 등 블록버스터 의약품들이 곧 제네릭 경쟁에 노출될 것임
<그림> 전세계 제네릭시장과 미국의 비중
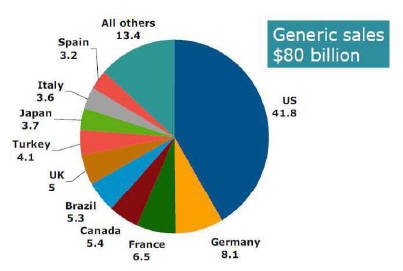
Source : IMS Health, MIDAS, Market Segrnentation, MAT Jun 2009 Rx Only
○ 2013년까지 특허가 만료되는 블록버스터 의약품의 전 세계 시장규모는 1,370억 불에 이르며, 브랜드 약품 특허만료에 따라 미국에서만 2003년부터 2013년까지 910억 불에 달하는 새로운 제네릭 시장이 형성
<그림> 세계 상위 20대 제약사의 특허만료에 따른 제네릭 경쟁 현황
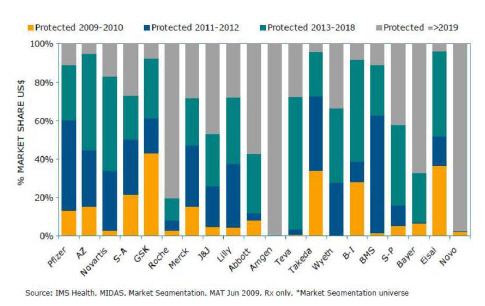
○ 아래 그림과 같이 현재 글로벌 제약사들이 제네릭산업 진출을 통해 구사하고 있는 다각화 전략을 이 같은 시장흐름의 방향성을 극명하게 보여주는 지표역할 수행
<그림> 매출부진을 만회하기 다국적 혁신신약 기업들의 제네릭 전략3)
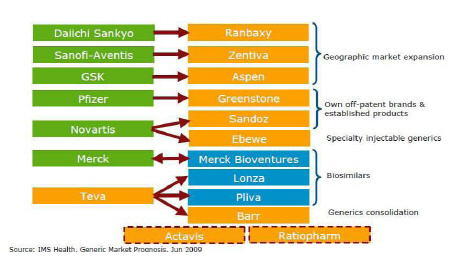
○ 혁신신약 기업들도 제네릭 분야의 관계사 진출 형태를 통해 연결 제무제표 상의 매출 격감 폭이 감소되고 있음
- 주주수익성 확보를 위한 경영합리화 추진으로 자체 연구 및 생산에 점차 보수적으로 변화되고 있으며, 제품의 Life Cycle을 유지하기 위해 개량신약(IMD, Incremen-tally Modified Drug)에 대한 인라이센싱을 적극화하고 있음
○ 2014년부터 건강보험 수혜자를 크게 확대시키는 미국 건보개혁은 이러한 제네릭 의약품 증가추세와 맞물려 거대한 제네릭 시장 확장 효과를 창출할 것으로 기대
○ 현재 미국 FDA에 허가 신청되는 의약품의 70%가 제네릭이며, 2009년 Harris poll에 의하면 미국인의 80% 이상이 제네릭을 선호한다고 조사
· 다국적 기업들 역시 지난 수년 사이에 제네릭 시장에 뛰어 들면서 에버그린(evergreen) 정책을 펼치고 있어 제네릭 경쟁은 치열해지고 있음
○ 반면, 그간 국내 제네릭의약품의 미국진출 실적은 거의 전무한 상황이며, 진출을 어렵게 한 다양한 요인들이 분석된 바 있음
- 유통라인 미확보, 제조물책임법에 의한 배상 가능성, 비교우위 제네릭 품목 미확보, 가격경쟁력 미흡, 특허정보 미흡 등 불확실 요인 상존
- 미국의 제네릭의약품 생산방식은 단일라인, 단일제품의 대량생산체제이나 국내는 소량생산 방식으로 미국 FDA 현장 실태조사 부담
○ 미국 내 시장점유율이 높은 인도, 중국산 제네릭의약품의 가장 큰 강점은 가격경쟁력임
- 이는 인건비 등 상기 국가의 비용구조에 기인하여, 일반적으로 오리지널 의약품의 특허만료 이후 평균 90% 가까이 가격이 격감하는 미국 제네릭시장의 가격형성을 주도해 왔음
- 또한 총매출액에서 매출원가를 제한 매출총이익(Gross Margin)의 경우도 제네릭의 경우는 오리지널 산약(80%)을 훨씬 밑도는 40%에 불과
○ 그러나 다국적 제약사의 기술이전 트렌드와 마찬가지로 제네릭의약품의 경우도 1차 치료(Primary care)의약품에서특수질환 의약품(Specialty)으로 전환해야 함
- 특수질환 제네릭의약품의 기술필요에 따라 가격경쟁력에 의해 대량생산시장을 선점하고 있는 인도와 중국은 이 분야에 경쟁력이 높지 않음. 또한 특수질환제 제네릭 의약품의 매출총이익(Gross Margin)은 약 60%로 일반 제네릭 의약품의 가격을 크게 상회함
○ 또한 의약품 성분혼합(formulation combination) 혹은 제형개략에 따른 개량의약품의 경우가 전망이 밝을 것으로 예상
· 고지혈증 치료제 Lipitor와 혈압강하게 Norvasc를 결합한 Pfizer의 Caduet 같은 제품
- 그러나 이를 위해서는 지속저인 연구개발 능력의 강화가 필요하며, 국내위주의 품목개발에 치우쳐 선진국 시장 내 지적재산권 관련 기술 및 노하우가 부족한 점, 국내에서 진행한 생물학적 동등성 실험과 임상데이터는 활용가치가 적으며 현지 영업과 사업능력은 전무한 것이 문제점임
○ 따라서, 한국의 제네릭의약품 기업들은 우선 생산 공장의 프로세스 중심, 파이프라인 중심으로 경쟁력을 활보한 후 공급시장에 도전할 수 있으므로 중장기적인 기업 내 혁신활동 추진이 병행되어야 함
- 중요한 성공요소는 보유한 제품화 기술을 통해 팔릴 수 있는 개량신약, 퍼스트 제네릭(first to file) 창출이 필수적이며,
- 생산 아웃소싱 유치를 실현할 수 있는 cGMP 수준의 품질경영 필요
- 미국 내 업계동향, 업체별 수요를 평가할 수 있는 미국 현지조직 구축 필요
○ 미국 기준인 cGMP 생산시설 구축과 해외마케팅 능력배양 등 제약 산업의 수출전략 산업화를 위한 정부와 산업계의 노력이 강화되고 있어 우수사례를 만들어낸다면 제네릭의약품 수출을 위한 물꼬를 틀 수 있을 것으로 예상
□ 영바이오의약품 및 바이오시밀러 제품 진출 가능성 확대
○ 이번 건보개혁에 포함된 생물의약품의 갸격 경쟁 및 혁신법(Biologics Price Competition and Innovation Act of 2009)은 그간 업계와 전문가를 중심으로 논의되어 오고, 의회에서 제안되어 왔던 법안을 보건의료개혁법에 포함하여 제정한 것임
- 동등생물의약품의 시장출시를 지연시키고 출시 후에는 대체처방을 불인정함으로써 오리지널 약품의 개발촉징과 브랜드 파워를 유지하는 전략을 모색해왔던 미국바이오협회(BIO, Biotechnology Industry Organization) 및 미국제약협회(PhRMA) 등 바이오 제약업계는 본 개혁내용을 적극 환영하고 있으며, 미국 의료계(AMA)도 지지 입장4)
- 반면, 고가의 의약품을 포함한 과중한 의료비 부담을 고민하는 정부와 제네릭 의약품업계를 대표하는 미국 제네릭의약품협회(GPhA)는 동등생물의약품의 승인 촉진을 통해 가격 경쟁 및 소비자의 접근성 향상을 추진해 왔으며, 본 바이오개혁은 건보개혁의 목적에 반하는 조치로 평가5)하고 미국은퇴자연맹(AARP)도 값싼 동등생물의약품의 출시 지연을 우려
○ 현재 제약 산업은 기존의 합성의약품에서 생물의약품으로 산업의 중심이 이동6)하고 있으며, 이번 조치는 생물의약품 및 동등생물의약품의 시장에 큰 영향을 미칠 것으로 전망
- 특히 Promoting Biologics Price Competition and Innovation이 전략으로 포함된 이번 건보개혁을 통해 동등생물의약품 제조를 위한 새로운 FDA 가이드라인7) 마련 및 동등생물의약품에 대한 친시장적 환경이 조성될 것으로 예측됨
· 동등생물의약품 허가 신청자의 수수료(User fee) 부과기준도 ‘12.10까지 마련토록 규정
○ 이 같은 변화는 현재 정부와 산업 공동으로 동등생물의약품 산업을 미래성장동력으로 설정하고 발전 계획을 수립하고 있는 우리나라에 기회를 제공할 전망
- 현재 바이오시밀러 개발에 관여되어 있는 국가는 EU(유럽), 호주, 일본, 말레이시아, 싱가포르, 한국, 대만(아시아), 콜럼비아, 멕시코(남미), 요르단, 쿠웨이트, 터키(중동), 캐나다, 미국(북미) 등으로 매우 제한되어 있음
- 이는 바이오의약품은 합성의약품과 달리 높은 개발비용 및 기술력이 요구되기 때문임
- 동등생물의약품 제품들은 보통 개발에 8~10년의 기간이 소요되며, 최종 제품 출시까지 약 1억~2억 달러 정도의 비용이 소요되는 것으로 추산
· 생물의약품제품에 높은 개발비용이 소요되는 것은 제조, 임상실험, 시판 후 조사 등에 고비용이 발생하기 때문이며, 이와 대조적으로 합성의약품 제네릭 개발비용은 약 1백만~5백만 달러 정도로 추산(IMS Health)
<그림> 동등생물의약품 생산에 필요한 임상절차
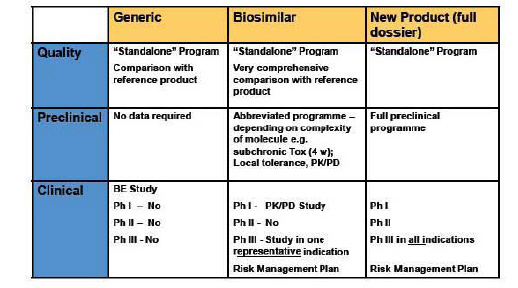
Source : Johnson&Johnson, 2009
○ 또한 일반 합성의약품이 약국 등의 유통경로를 통해 최종 소비자인 (end user) 환자에게 직접 전달되는 반면, 바이오의약품은 대부분 보건의료 제공자(의사)를 통해 ‘ 의료식 치료’의 하나로 환자들에 전달되어 합성의약품의 시장점유율이 큰 영향을 미치지 못하는 새로운 경쟁 영역임
○ 현재 미국의 동등생물의약품의 시장과 규제기준이 형성단계인 만큼 정부의 적극적인 지원을 통해 바이오시밀러 개발에 집중하는 전략이 필요할 것으로 전망
- 특히 미국 FDA도 유럽의약청(EMEA)의 가이드라인을 참조할 것으로 보이기 때문에 EMEA의 승인에 성공한 LG생명과학의 사례 등은 우리기업의 개별 전략 수립에 준거로 활용될 전망
· 현재 미국 동등생물의약품 허가규정은 생물의약품 허가승인규정인 Public research service Act의 BLA(Biologics License Application)인 Section에 (K)항을 신설하는 형태로 입법 예상. 새로운 규정의 발효 전까지 생물의약품은 잠정적으로 합성의약품의 개량신약 승인규정의 허가루트를 거칠 것으로 보임
○ 우리나라의 경우 현재 정부 차원에서 바이오시밀러 제품화 및 세계진출 지원을 선언함에 있으며, 바이오신약개발을 위한 신성장 동력 핵심 연구개발 과제 역시 관계 부처별로 추진 중에 있음
- 바이오 분야의 근거법인 생명공학육성법 역시 기존의 연구개발 중심에서 사업화 촉진을 위한 패러다임으로 전환하고자 계획하고 있고, 다양한 육성프로젝트를 진행하는 등 집중 지원을 모색하고 있음
○ 또한 현재 국내 기업들 역시 바이오시밀러를 성장 동력으로 인식하고 집중적인 투자와 연구개발을 시행하고 있으며, 다양한 공동개발, 공동연구를 통해 경쟁력 보유를 추진하고 있음
- 현재 경쟁력을 갖추고 있는 국내 바이오시밀러 기업으로는 인터페론, 서방형 신장호르몬의 개발을 성공한 LG생명과학, 2011년 상용화를 목표로 5년간 5,000억 원 투자계획을 수립한 삼성정자
- 바이오시밀러 ‘히셉틴’의 사업화가 완성단계에 와있는 것은 물론, 최근 종합 독감항체 치료제 개발에 세계 최초로 성공한 셀트리온
- 세포배양 방식의 인플루엔자 백신 개발 핵심기술인 세포주 확립에 성공한 녹십자, 바이오신약 부분에서 총 9개 신약을 개발 중인 동아제약 등 있음
○ 특히 우리 기업들의 경우 생산기술의 효율성와 안정성이 강점이어서 핵심 플랫폼 기술을 보완해 줄 파트너사를 확보한다면 바이오시밀러 시장에 경쟁력이 있을 것으로 보임
- 미국이 오리지널 바이오의약품에 12년 독점권을 부여한 것은 향후 의약산업의 핵심은 게놈, 단백체(Genome, Proteomics) 등 바이오의약품이라는 것을 선언한 것으로 해석이 가능
- 지금까지 바이오테크를 개발하기 위한 국내의 수많은 노력이 있었기 때문에 전통적인 합성의약품과의 적절한 협력을 통해 유리한 위치 선점 가능
- 이를 위해서는 정부의 지원형태는 기업과 기업의 기술을 중개하고 연결해주는 파트너링 위주의 행사 지원을 넘어 제도적인 진출지원 체계 구축 시급
□ 백신 등 조기건강모델 (Early Health Model) 제품 수출 기회 확대
○ 미국 건보개혁법안은 국민들의 질병예방과 건강관리 증진 프로그램 가화를 정책적으로 장려
○ 현 법안에 따른 질병예방과 건강관리는 크게 4가지 방향으로 추진
- 국립 질병예방 및 건강증진 위원회 설치하고 관련 사업에 2015년까지 향후 5년간 70억 달러, 2015년 이후에는 매년 20억 달러 예산 편성
- 메디케어와 메디케이드 가입자 대상, 질병예방 및 건강관리 프로그램 확대
- 종업원에 건강관리 프로그램을 제공하는 중소기업에게 최대 5년 까지 정부보조금 지원 및 프로그램에 참여직원에 비용지급 등 인센티브 부여
- 체인점, 자동판매기 등의 음식에 영양성분 표기를 의무화
○ 미국의 경우 만성질환에 의한 사망률이 높아 예방의학과 건강관리는 향후 더욱 중요한 이슈로 지속적으로 부각될 전망
<그림> 미국이 주요 사망원인 (2005)
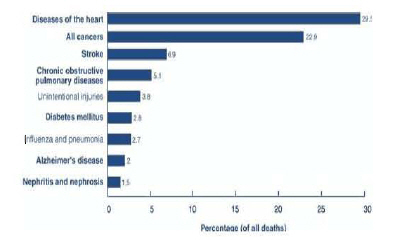
Source : Causes of Death USA, National Vital Statistics Report
- 세계보건기구(WHO)도 매년 발표하는 회원국의 사망원인 (Ten Leading Causes of Death, WHO)을 통해 만성질환을 향후 15년간 전 세계 사망원인의 3/4을 차지하고 심각한 장애를 유발하여, 종국적으로 자원 낭비와 경제성장과 발전을 저해할 위험인자로 꼽고 있음
○ 이와 같은 다방면의 질병예방과 건강관리 정책에 따른 관련 제품시장의 성장이 예상
- 특히 전통 한의학 등을 접목한 건강증진 제품과 웰빙 기능성 식품 등 미국제품과 차별화된 제품군을 보유한 국내 기업의 경우 진출 가능성 높음
- 혈압계, 당뇨측정기(glucometer), 진단기기, 진단시약 등 예방관리 차원에서 소요되는 의약품, 의료기기 등의 수요도 장기적으로 확대될 전망임
- 국내 금연보조제품(smoking cessation)의 시장 역시 연간 200억 원 수준으로 한독약품, 녹십자, 한미약품 등에서 다양한 금연보조제품을 생산아고 있어 미국시장으로 진출 확대가능성이 있음
○ 또한 신종 플루를 포함한 신형 전염성 인플루엔자가 유행함에 따라 이를 예방하기 위한 백신시장이 성장할 것으로 전망
- 타미플루 등 합성의약품의 치료 한계가 명확히 드러나면서, 바이오의약품인 백신에 대한 의존도가 높아지고 있음
- 국내의 대표적인 백신기업 녹십자가 신종플루 특수로 매출이 급증하면서 2010년 국내 2위 제약사로 도약하는 등 백신의 시장성이 확인되면서 연구개발에 나서는 업체들이 증가할 것으로 전망됨
○ 특히 유엔개발계획(UNDP)이 설립한 국제백신연구소(IVI)가 국내에 본부를 두고 있고 백신개발 연구의 선구자 역할을 한다는 것은 국내 산업에 긍정적인 인프라로 기능할 수 있음
- 국제백신연구소는 임상 면역학, 점막 면역학, 분자 미생물학, 분자 백신학, 백신 공정개발 및 기술이전 분야의 실험실 연구 프로그램 확대를 강화하고 있으며,
- 신종 바이러스를 포함한 병원체의 특성을 분석하고 대표 균주(strain)에서 백신용 후보 항원을 추출하기 위한 분자미생물학 플랫폼을 개설하는 등 다양한 혁신적 연구를 추진하고 있음
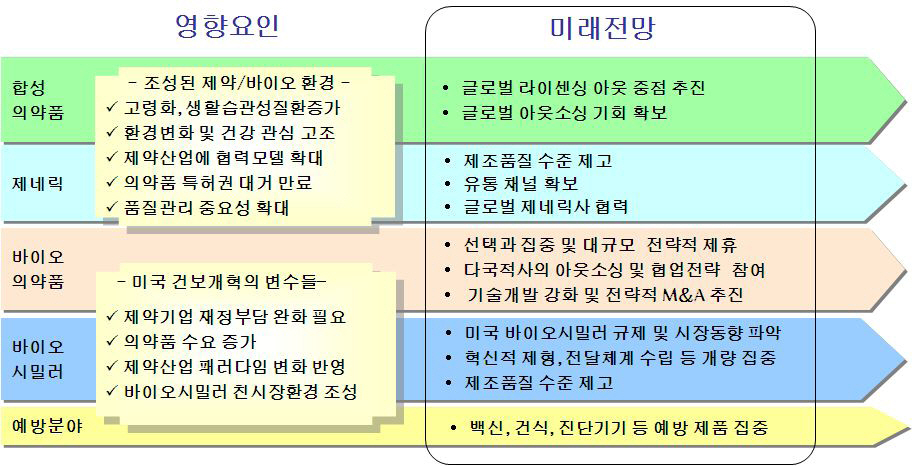
□ 긍/부정적 요인

□ 우리나라 바이오/제약에 미치는 영향 및 미래 전망 요약
1) cGMP(current GMP, 현행 우수제조관리기준) : 미 FDA의 관리기준
2) 제조공정·시설이 올바르게 작도하고 있는지를 체계적으로 조사·검토·확인하여 문서화
3) Ratiopharm은 2010년 3월 18일자로 Teva에 인수되었음
4) 그러나 의사들로 하여금 동등생물의약품의 오리지널 의약품에 대한 약효 동등성의 인정과 이러한 논리로 대체처방을 강제해서는 안 된다는 입장 표명
5) 5년의 독점긱ㄴ을 요구해온 GPhA(Kathlcen Jacger 회장)은 동법이 경쟁의 촉진과 소비자의 선택의 확대 등 접근성 제고에 실패했다는 입장임
6) 2015년까지 약 85%의 기존 오리지널 합성의약품의 특허기간 종료될 예정임
7) FDA는 법에서 규정하고 있는 biosimilarity 및 interchangeability의 의미에 대한 해석과 운용기준, 동등생물의약품의 허가 신청, 심사 및 승인 기준, 이에 필요한 임상시험 등의 자료제출 요건 등 업계를 위한 가이드라인을 설정 예상
보관함 담기












